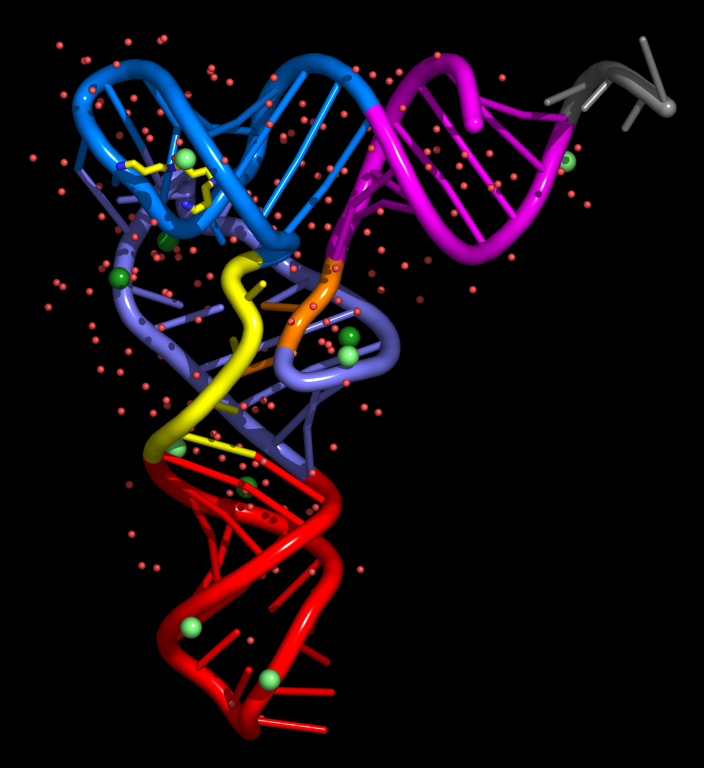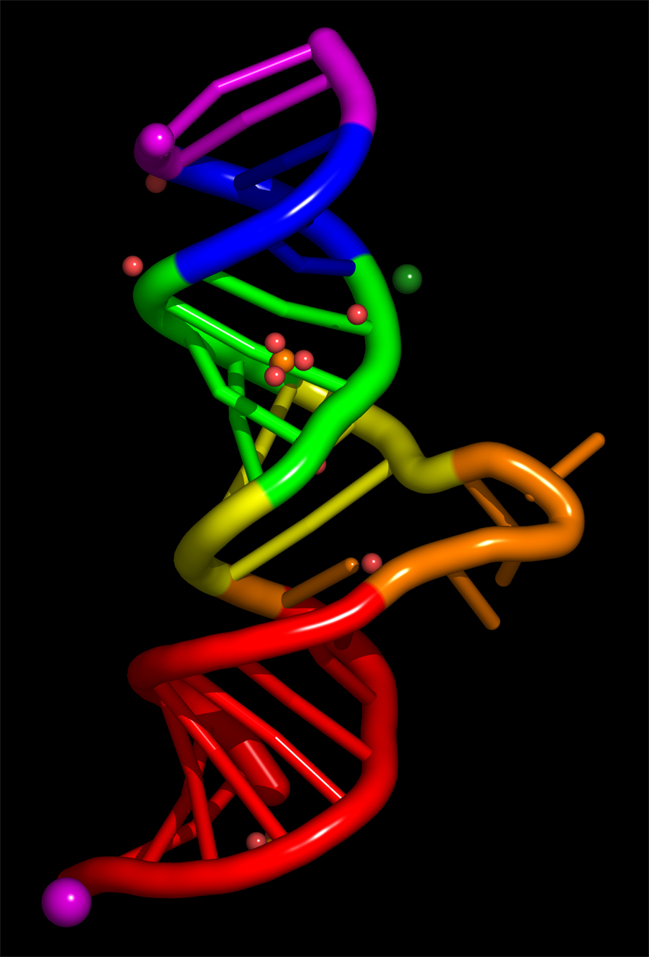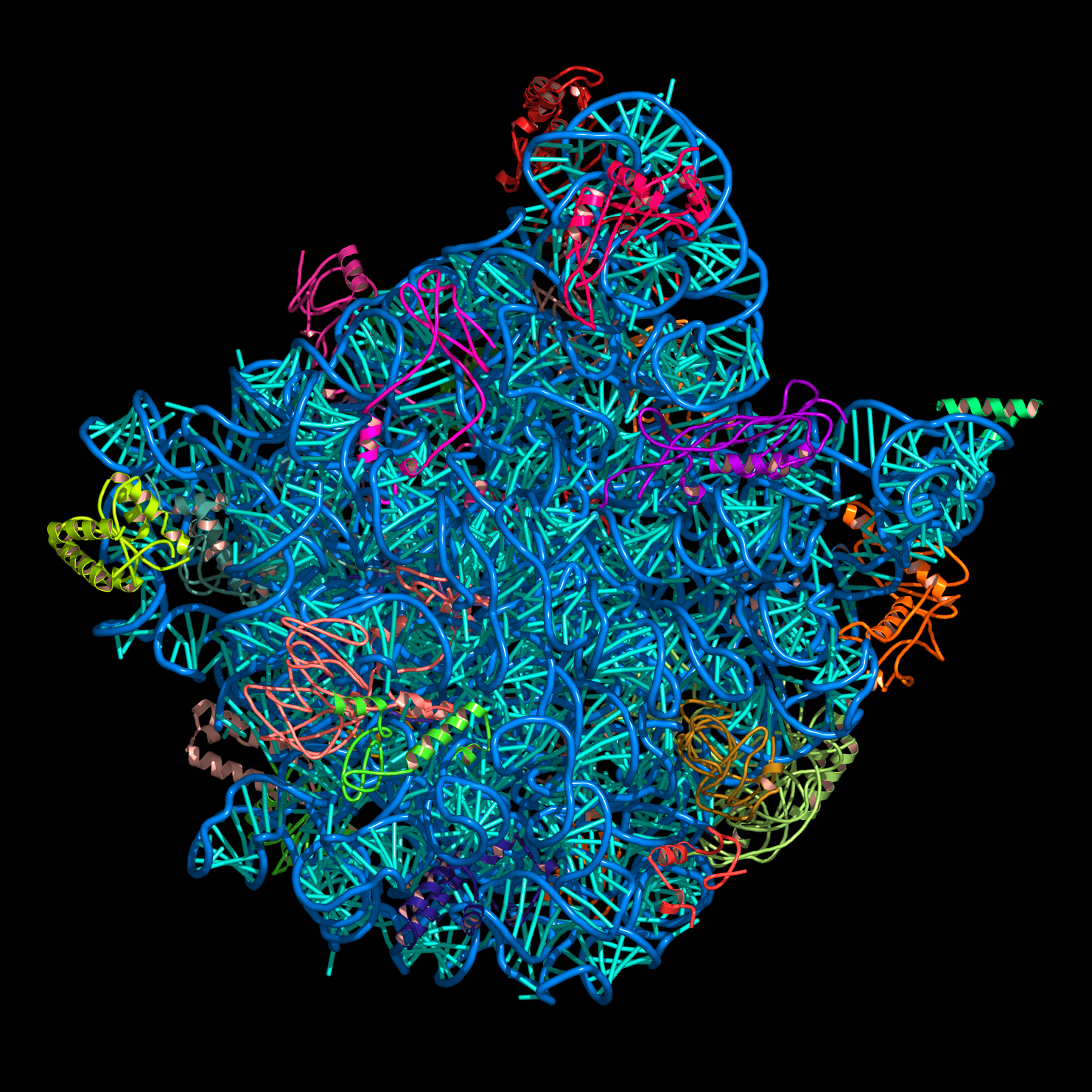
< click on the images for full size versions >
nuccyl version: 1.5.2
|
|
New in version 1.5.2:
- bug fix: nuccyl now processes correctly nucleic acids with palindromic sequences, or containing nucleotides with identical type and name belonging to different chains
- chain name information has been added to the name of cylindrical objects created by the "nuccyl -cyl_3" command: cylinder name syntax is now chain_typebase (i.e. the cylinder representing residue U8 within chain A is now called A_U8 instead of just U8)New in version 1.5.1:
- support for additional modified nucleotides (AET, G7M)
New in version 1.5:
- filled base representation of nucleic acids
- support for all modified purine and pyrimidine nucleotides currently in PDB
- run length printoutIMPORTANT: due to the introduction of the new nuccyl -filled command, commands nuccyl -1 / -2d / -2 d / -3 have been renamed nuccyl -cyl_1 / -cyl_2d / -cyl_2d / -cyl_3 for clarity!
nuccyl is a Perl program that allows PyMOL, a powerful open-source molecular modeling system, to display atomic models of nucleic acids in a highly simplified representation. By depicting the nucleic acid backbone as a cartoon and the bases of nucleotides as cylinders, similarly to the excellent programs Ribbons and Drawna, nuccyl and PyMOL allow to quickly grasp the overall fold of an RNA or DNA molecule:
< click on the images for full size versions >
nuccyl produces base cylinder coordinates and command files to display them with PyMOL. The base pair analysis programs RNAView and 3DNA can also be optionally used to obtain starting input files for nuccyl.
You can find information about PyMOL and download the program here. This document assumes that you have already installed PyMOL and are already familiar with the program - please refer to the program's manuals, FAQ and user pages.
RNAView can be downloaded and installed as described here; a publication describing the program is also available.
Information and download links for 3DNA may be found at the program's home page; a paper has also been published.
nuccyl was developed and tested on a PowerBook G4 1.5 GHz with 2 GB of RAM (running OS X 10.3.8). Should you have comments and/or problems with your particular setup, please contact me at so I can update this document.
Test before you try... |
|
Before downloading nuccyl and learning how to use it, you might want to have a look at these PyMOL session files, which I used to generate the images shown above:
45s_rna.pse.gz
50s.pse.gz
p4p6.pse.gzAfter decompressing any of these files:
you can recreate the corresponding scene by loading the session into PyMOL (version 0.98+ required):
This should give you an idea of the kind of representations that you will be able to get from the combination of nuccyl and PyMOL.
Part I - nuccyl installation |
|
nuccyl is an OSI certified unrestricted open-source program. Before you download nuccyl, please read its license. Then download the software by clicking on the following link:
Uncompress and extract the files in the archive:
UNIX> tar xvf nuccyl_1.5.2.tarAfter moving the resulting folder (nuccyl) to a directory of your choice (for example, ~/bin), make sure the program can be executed:
UNIX> chmod 755 ~/bin/nuccyl/nuccyl.plNote that, if the Perl interpreter is not installed on your system as /usr/bin/perl, you will need to edit the first line of nuccyl.pl so that it includes the path to your Perl installation.
Now type:
which should return:
I will now describe a real-life example of how to use nuccyl, together with RNAView and PyMOL, to display the structure of yeast tRNAPhe as shown above (if you are working on a DNA molecule, you should still go through the whole tutorial since most of the steps are identical). Please keep in mind that, although the procedure might at first seem somewhat complicated, in practical terms it is very fast - unless you work on the ribosome, it shouldn't take you more than 15-30 minutes to apply it to your own cases!
First of all, make a new directory called nuccyl_1evv:
and copy the nuccyl executable into it:
I highly recommend making a separate directory for every project you use nuccyl for, as well as including your current nuccyl.pl executable in each of these directories. This is because some of the intermediate files created by nuccyl have standard names (so that subsequent runs will overwrite previous ones), and it will allow to keep track of which version of the program you actually used for each particular run.
Now download the yeast tRNAPhe coordinates from PDB:
UNIX> cd ~/Desktop/nuccyl_1evv
IMPORTANT: if your starting PDB file does not come from PDB, please make sure that it is formatted as described on the RCSB web site. (you can easily reformat PDB files created with CNS and other programs using CCP4's pdbset and coordconv or Gerard Kleywegt's ACONIO). In particular, nuccyl assumes that all your nucleotides will have a chain identifier - strange things will happen if they don't.
Step 1 - Process modified nucleotides |
||
If your PDB file does not contains coordinates for modified nucleotides, skip to Step 2 (however, for the purpose of this tutorial, you might still want to go through Step 1 - if nuccyl will not detect any modified nucleotide, it will simply create identical copies of your PDB file to be used during subsequent steps). Otherwise - as in this tRNAPhe example - you will need to edit the coordinate section of the modified nucleotides in order for RNAView and PyMOL to properly work with them.
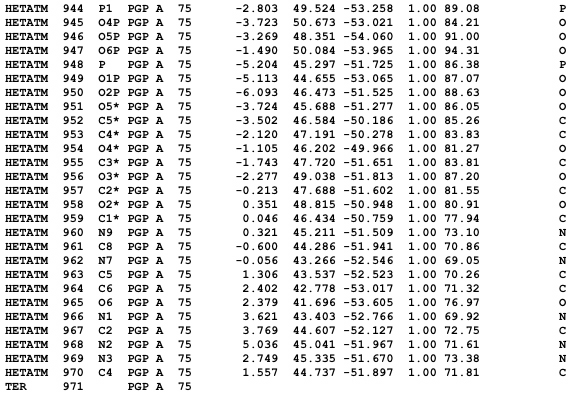
First of all, if any of your nucleic acid chains ends with a 3' cyclic phosphate (which is not the case for tRNAPhe), such as:
You will need to manually edit it in the PDB file as follows:
This will redefine the cyclic phosphate group of the 3' nucleotide (nt 75 in the example above) as the phosphate group of an extra nucleotide (nt 76). Although the rest of this newly defined nucleotide is missing, its phosphate group will be enough to tell PyMOL to extend the nucleic acid backbone cartoon to the very end of your chain; otherwise, in the example above, the cartoon would stop prematurely.
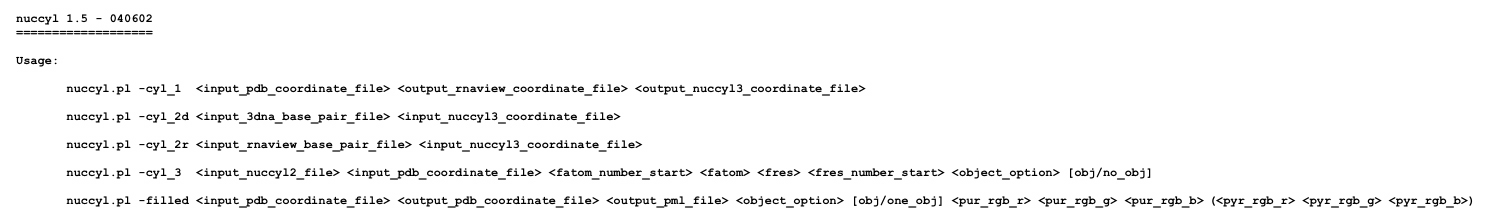
Next, you will need to take care of any other type of modified nucleotide that you might have in the structure. You can do so automatically and generate modified PDB files for RNAView and PyMOL by running nuccyl with a -cyl_1 flag (you can get information about the command syntax by simply typing "./nuccyl.pl -cyl_1"):
The above command will generate: (1) a PDB file for RNAView (1EVV_rnaview.pdb), where HETATM has been converted to ATOM at the beginning of each modified nucleotide coordinate line and modified nucleotide names have been converted to their corresponding unmodified counterparts (i.e. 1MA
A, etc.) and (2) a PDB file for nuccyl itself and PyMOL (1EVV_nuccyl3.pdb), where HETATM has been converted to ATOM at the beginning of each modified nucleotide coordinate line.
In both files, actual atomic coordinates are NOT modified.
Note that, in most cases, one does not need to create a modified PDB file for PyMOL, since the program can deal with modified nucleotides by using the commands:
PyMOL> sort
PyMOL> rebuildHowever, in my experience these commands can occasionally lead to missing bonds between O3' and P atoms, which does not happen if the PDB file is externally pre-edited by nuccyl.
The current version of nuccyl recognizes all modified nucleotides currently in PDB; however, should your PDB file contains additional ones, please so that I can add the missing specifications to the nuccyl distribution.
For RNA molecules, the easiest way to quickly prepare a starting input file for nuccyl is to analyze your PDB file with RNAView (alternatively, you can use 3DNA, as described below for DNA molecules).
In our case:
Now you can convert the output from RNAView (file 1EVV_rnaview.pdb_sort.out) to a simpler format by running nuccyl with a -cyl_2r flag (again, you can get information about the command syntax by simply typing "./nuccyl.pl -cyl_2r"):
(note that the second input file specified above, 1EVV_nuccyl3.pdb, was one of the two files generated during Step 1; here, nuccyl uses this file to retrieve the original names of modified nucleotides (i.e. 1MA instead of A, and so on...) which were replaced with standard ones in order for RNAView to work).
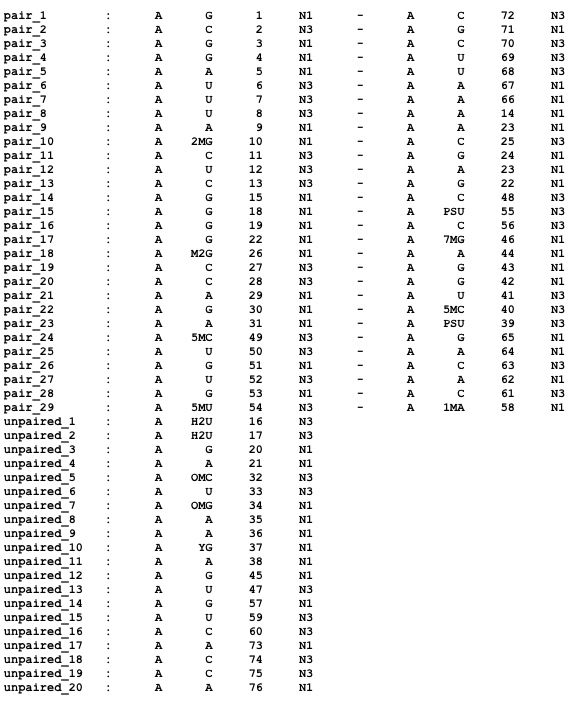
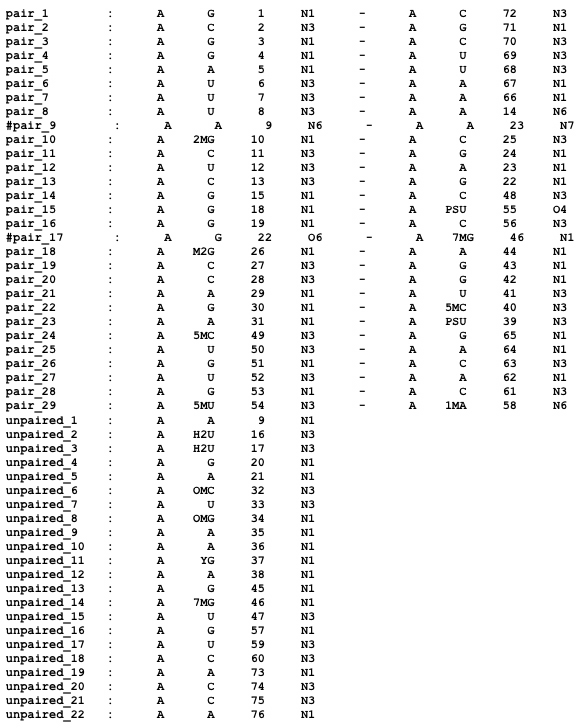
For each base pair identified by RNAView, the output file from a nuccyl.pl -cyl_2r run (nuccyl2.out) contains this information:
and, for each unpaired nucleotide, the following:
In the case of tRNAPhe, this corresponds to:
nuccyl will use the information to calculate the intersections between the cylinders representing the various base pairs, which will be displayed by PyMOL.
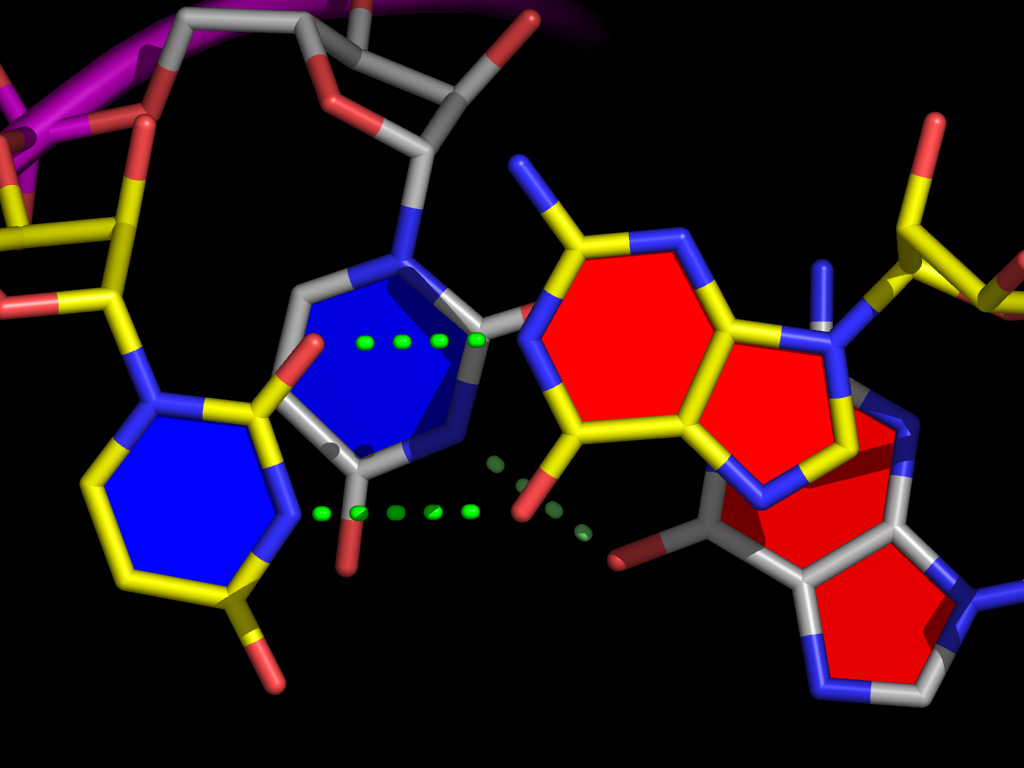
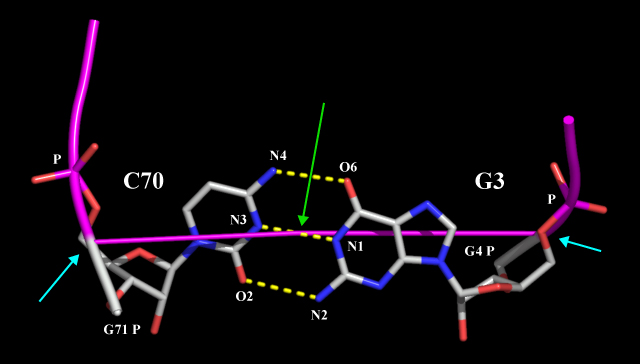
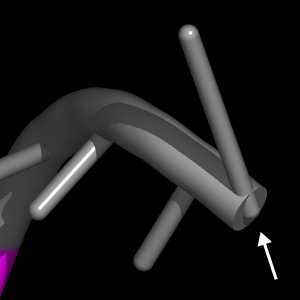
For example, the data on the line for pair_3 above tells nuccyl to use the middle point between the N1 atom of G3 (chain A) and the N3 atom of C70 (also in chain A) as the coordinate (indicated by a green arrow) where the cylinders representing the two bases will meet to indicate their pairing:
Note that nuccyl will automatically use the middle point between the P atom of a given nucleotide (N) and the P atom of the nucleotide that follows it (N+1) as the starting point for the cylinder representing the base of the nucleotide N itself (cyan arrows).
If the nucleotide at the 5' end of a given nucleic acid chain lacks the phosphate group, nuccyl will use its C5* instead of the P atom.
On the other hand, the starting point for the cylinder representing the base of the nucleotide at the 3' end of a chain defaults to the P atom of the 3' nucleotide itself. This is because nucleic acid backbone cartoons drawn by PyMOL end precisely at this atom, so any other choice would cause the 3' base not to be connected to the backbone.
The nuccyl2.out file can be manually edited to:- comment out (with "#") base pairs that you do not wish to show (for example A9-A23, which generates a base triple)
- add extra unpaired nucleotides that you also wish to show as cylinders (for example the "third" nucleotide involved in a base triple that you may have commented out, as just described above)
- change the base atoms that are used to draw the cylinders representing nucleotide pairs (see below)For example, my final tRNAPhe input file for nuccyl (nuccyl2.out_edit), derived from nuccyl2.out, looks like this:
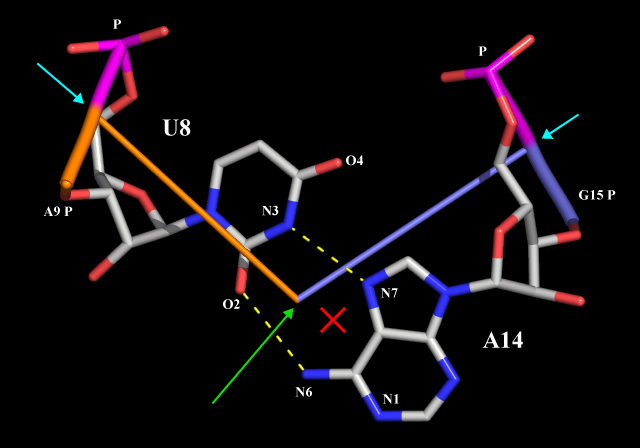
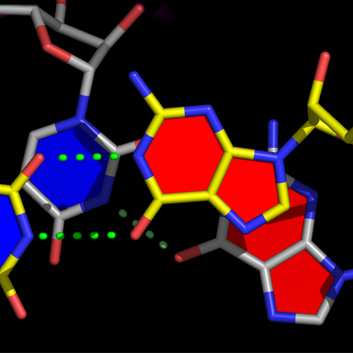
You can see that I have manually modified some of the base atoms from their defaults (N1 for purines and N3 for pyrimidines). The reason for this is obvious if one considers a base pair like U8-A14:
In a case like this, using the N1 atom of A14 to define the "center" of the base pair would clearly not be a good choice - the base cylinders would intersect at the position of the red cross. A more satisfactory result can on the other hand be obtained by specifying that N6 of A14 should instead be used to calculate the base pair cylinder intersection. Thus, by manually editing nuccyl2.out files, you can gain complete control on what nuccyl will tell PyMOL to display.
For DNA molecules, you can quickly prepare a starting input file for nuccyl by analyzing your PDB file with the program find_pair of the 3DNA package. For a generic PDB file dna.pdb this can be done as follows:
You can then convert the output from find_pair (file dna.inp) to a simpler format by running nuccyl with a -cyl_2d flag (once more, you can get information about the command syntax by simply typing "./nuccyl.pl -cyl_2d"):
The next step is to tell nuccyl to read the (edited) base specifications and generate cylinder information for PyMOL. nuccyl will (1) output a PDB file containing coordinates of pseudo atoms representing the start and end points of each cylinder (file nuccyl3.pdb), and (2) ouput a PyMOL command file for displaying the cylinders (file nuccyl3.pml).
To do so, invoke nuccyl with a -cyl_3 flag (once more, you can get information about the command syntax by simply typing "./nuccyl.pl -cyl_3"):
the arguments following the -cyl_3 flag being:
- the name of the (edited) base specification file created in Step 2 (nuccyl2.out_edit)
- the name of the formatted coordinate file created in Step 1 (1EVV_nuccyl3.pdb)
- a starting atom number for the pseudo atoms used to draw cylinders (5000)
- an atom name for the pseudo atoms (B)
- a chain identifier for the pseudo atoms (X)
- a starting residue number for the pseudo atoms (500)
- a flag specifying whether to create a PyMOL object for each base cylinder output by nuccyl (obj), or not (no_obj)You can follow what nuccyl is doing both interactively and by looking at its full log file (nuccyl3.log). The program also outputs a file called nuccyl3.problems, in which all warnings and problems that were automatically fixed by nuccyl are logged. Make sure that you go through the latter, especially if what ends up being displayed by PyMOL in Step 4 does not look like what you expected!
In our example case, the beginning of output file nuccyl3.pdb looks as follows:
and the corresponding section of output file nuccyl3.pml is:
These specific lines tell PyMOL how to display the cylinders corresponding to base pair G1-C72; note that each cylinder is defined as an object having the same name of the base it represents, because I used the obj flag when I issued the nuccyl -cyl_3 command above.
Concerning this point, keep in mind that creating objects within PyMOL is currently very slow. Therefore, if your structure consists of a large number of nucleotides (like the 50S ribosomal subunit shown above), you should definitely consider running nuccyl -cyl_3 with a no_obj flag. In this case, the output nuccyl3.pdb file will be identical to the one you would obtain using the obj flag, but the nuccyl3.pml file will instead have all lines starting with create commented out. As a result, when PyMOL will read the nuccyl3.pml file, it will still create bonds between pseudoatoms (i.e. cylinders), but will not generate corresponding objects. As you can easily find out by comparing runs with obj and no_obj flags, this will speed up the procedure tremendously.
At first, it might seem very inconvenient not to generate objects corresponding to the various bases, since these can be easily manipulated using PyMOL's internal GUI (see picture below). However, currently PyMOL does not allow to sort the list of objects within the internal GUI itself, so that the advantage of using the latter becomes negligible if one is working with a large number of nucleotides - unless they enjoy scrolling thorugh a semi-random list of hundreds of nucleotides, that is.
Why does nuccyl bother to write create... lines to its output file nuccyl3.pml, if these lines are commented out (and hence not read by PyMOL) when using a no_obj flag? The reason is that this information can still be very useful if one wants, for example, to assign different colors to various regions of an RNA molecule; this is because, by inspecting the nuccyl3.pml file, one is able to immediately find out which pseudoatoms/cylinders correspond to a given range of nucleotides.
We are finally ready to have a look at the results. Start PyMOL and load interactively the files created by nuccyl:
PyMOL> load 1EVV_nuccyl3.pdb, 1evv
PyMOL> load nuccyl3.pdb, nuccyl3
PyMOL> @nuccyl3.pml
then issue these commands:
PyMOL> cartoon loop
PyMOL> show cartoon, 1evv
PyMOL> zoom complete = 1
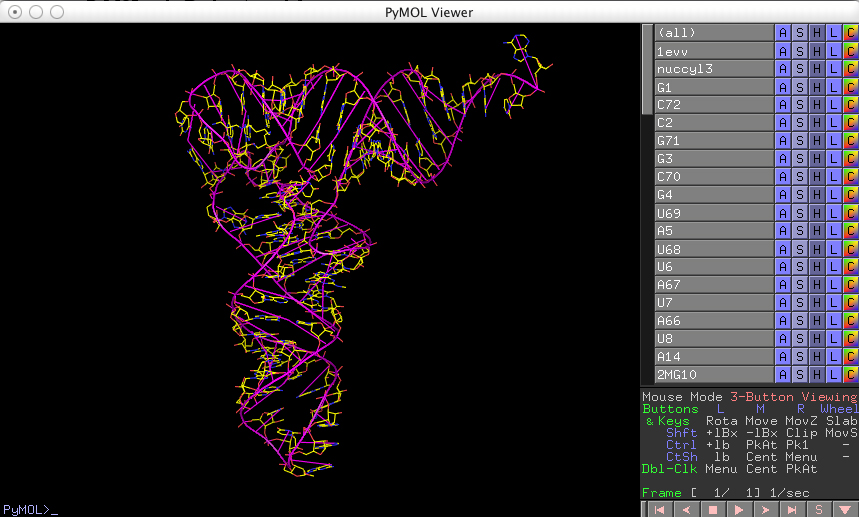
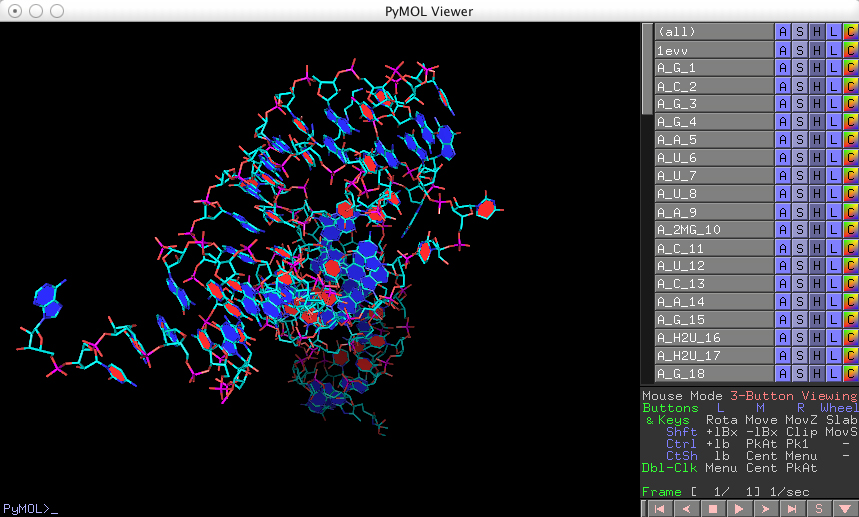
and you should see the following (after rotating the model):
Note, on the right hand side of the viewer window, the PyMOL internal GUI with a list of objects corresponding to the various base cylinders (listed in the order in which they were created).
You can now experiment with PyMOL's cartoon and stick settings to modify the backbone ribbon and cylinder properties. The particular settings I used to make the picture shown at the beginning of this documents are:
PyMOL> set cartoon_loop_quality = 25
PyMOL> set cartoon_refine_tips = 25
PyMOL> set cartoon_power = 2
PyMOL> set cartoon_power_b = 0.5
PyMOL> set cartoon_sampling = 20
PyMOL> set cartoon_throw = 1
PyMOL> set stick_radius = 0.4
In addition, you can modify individual cylinders by using the buttons to the right of each base within the internal GUI object list.
The ends of nucleic acid backbone cartoons displayed by PyMOL precisely coincide with the P atoms of the 5' and 3' nucleotides.
As a result, the cylinders representing the 5' and 3' bases of a nucleic acid chain stick out a bit from the backbone cartoon:
This issue can be tackled by displaying the extreme P atoms as spheres of an appropriate radius. In the case of tRNAPhe:These commands will round the chain ends:
PyMOL> color magenta, resi 1 and name p
PyMOL> show sphere, resi 1 and name p
PyMOL> set sphere_scale = 0.55, resi 76 and name p
PyMOL> color grey, resi 76 and name p
PyMOL> show sphere, resi 76 and name p
A second problem is that the cylinder representing the 5' base ends up being disconnected from the cartoon if the 5' nucleotide does not have a phosphate group:
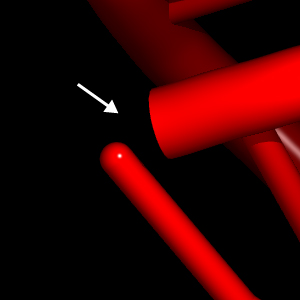
Missing 5' phosphates can also be dealt with - albeit less satisfactorily - by (1) specifying a bond between the O5* atom of the first nucleotide and the P atom of the second one, and (2) displaying the two atoms as spheres and the bond that connects them as a stick. Also in this case, you will have to experiment with different values of the PyMOL parameters sphere_scale and stick_radius to get a smooth transition between the various elements. For example, in the case shown above (5' end of 4.5S RNA domain IV), I used the following:
PyMOL> color red, ((resi 31 and name O5*) or (resi 32 and name P))
PyMOL> set stick_radius = 1.3, ((resi 31 and name O5*) or (resi 32 and name P))
PyMOL> show sticks, ((resi 31 and name O5*) or (resi 32 and name P))
PyMOL> set sphere_scale = 0.71, ((resi 31 and name O5*) or (resi 32 and name P))
PyMOL> show spheres, ((resi 31 and name O5*) or (resi 32 and name P))
which gave this result:
Once you have found a combination of commands and parameters that you like, you can automate the whole procedure by including them in a single PyMOL command (.pml) file. This is the one I used for tRNAPhe:
This file can be loaded into PyMOL by simply typing:
and will generate a PyMOL session file phe_trna.pse. Binary .pse files are much faster to load into PyMOL than command .pml ones; however, it is important to also keep the latter for reference, since they are invaluable for reconstructing the procedure that one followed to obtain a certain scene.
To create ray-traced pictures from a PyMOL session file, a command file like the following can be used:
Since version 1.5, nuccyl is also able to output information for PyMOL to display nucleic acids in filled base representation.
Here's how to do it:
To generate base filling information for PyMOL, all you need is a PDB file and a single nuccyl command. In this case, nuccyl must be invoked with a -filled flag (as usual, you can get information about the syntax of this command by just typing "./nuccyl.pl -filled").
Always using tRNAPhe as example:
The arguments that follow the -filled flag are:
- the name of the starting input PDB file (1EVV.pdb)
- the name of the output PDB file for PyMOL (1EVV_pymol.pdb)
- the name of the output command file for PyMOL (1EVV_filled.pml)
- a flag specifying whether to create a base filling object for each nucleotide (obj), or only two objects, corresponding to purine and pyrimidine base fillings, respectively (no_obj) (this is explained in more detail below)
- RGB values specifying the color of the base filling for purines (in this case, blue (0 0 255))
- RGB values specifying the color of the base filling for pyrimidines (in this case, red (255 0 0))Note that, if you only supply 3 numbers (i.e. a single RGB color) at the end of the argument list, nuccyl -filled will use the corresponding color for filling both purine and pyrimidine bases.
Start PyMOL and load interactively the files created by nuccyl:
PyMOL> load 1EVV_pymol.pdb, 1evv
PyMOL> @1EVV_filled.pml
then issue these commands:
PyMOL> set stick_radius = 0.15
PyMOL> show sticks
PyMOL> zoom complete = 1
and you should see the following:
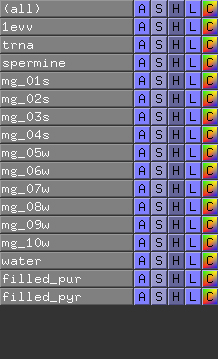
Base fillings are generated under the form of CGO objects. Because we used the obj flag as fourth argument to the nuccyl -filled command above, nuccyl output information for PyMOL to create an individual CGO object (i.e. base filling) for each nucleotide base, with names of the type X_Y_# (where X = chain identifier, Y = residue name and # = residue number). You can see a list of these objects on the right hand side of the viewer window, within PyMOL's internal GUI name panel.By clicking on the S and H buttons at the right hand side of the CGO object names, you will be able to either display or hide the filling of each individual base. This functionality clearly is handy, however - as explained before - object creation within PyMOL is slow, and if your nucleic acid model is large you might not want to generate an individual CGO object (i.e. base filling) for each of its nucleotides. To specify that you do not want individual base fillings, use the no_obj flag as fourth argument to the nuccyl -filled command; nuccyl will then instruct PyMOL to create only two CGO objects, one corresponding to the fillings of purine bases (filled_pur), and the other to those of pyrimidine bases (filled_pyr). As a result, although the representation of the molecular model will be identical to the one shown above, you will only see two CGO objects listed in PyMOL's internal GUI name panel:
What if you want to specify different filling colors for individual bases, or sets thereof? Just use your favorite editor to edit nuccyl -filled's output command file for PyMOL (1EVV_filled.pml in the example above), substituting overall RGB values with the ones you want to assign to specific residues.
For example, to change the base filling color for nucleotide G4 of chain A from blue to green, modify lines:
to:
(note that, in the case of purines, base filling is actually generated as the sum of two CGO objects, corresponding to the two rings of the purine base; in other words, in the example above A_G_4 = A_G_4_1 + A_G_4_2. Should you want to, you could take advantage of this implementation to specify different colors for the two rings of a given purine base)
After you are done with the editing, simply reload the modified command file into PyMOL:
Hopefully, the tutorials on this page explained clearly enough how to use nuccyl together with RNAView/3DNA and PyMOL. Should you want to have a look at the example files I discussed in this document, you can download them here:
Naturally, the best way to extend PyMOL is to write additional program modules in Python. However, my experience with nuccyl suggests that, by outputting a series of PyMOL commands, other programming languages can be just as effective. Using the same strategy, any user with some programming skills - and not just those who know Python - can immediately add new features to PyMOL. Thus, nuccyl will hopefully represent the prototype of a new series of tools that exploit an alternative way to improve PyMOL's functionality.
Updates |
||
You can find nuccyl's release history here.
If you would like me to let you know when a new version of nuccyl is released, please . Needless to say, I will also highly appreciate any comment, suggestion or criticism that you might have.
There is no paper describing nuccyl (yet). However, if you found the program useful and would like to mention it in your publications together with PyMOL, you could use an URL citation of the type:
Finally, I would also be very grateful if you could let me know when you publish a manuscript that includes figures made with nuccyl. This is because it would be useful to list these papers here, so that new users could have a better idea of what can be done with the program (other than the few examples I show above).
Thank you!
The following publications include figures made with nuccyl - when available, click on the thumbnails for full size pictures!
Acknowledgements |
||
Many thanks to Kevin Weeks for finding ways to display base pairs as cylinders within PyMOL and smoothen the ends of nucleic acid chains, and to Gerard Kleywegt for supplying me with a comprehensive list of modified nucleotides. Thank you also to Warren DeLano, Tassos Perrakis, Ken Westover, Jeff Chao, Peter Haebel, Xiang-Jun Lu, Tony Giannetti, David Jeruzalmi and Michal Jakob for their interest, comments and precious suggestions.